|

食品分析のさらなる向上を目指す
~定量NMRを用いた新たなアプローチ~ 日本大学 生物資源科学部 食品生命学科
准教授 松藤 寛 専任講師 大槻 崇 1.はじめに天然素材や食品から、何らかの生理活性物質を見出し、これを有効に利用することで、健康の維持や増進並びに高品質な食品の創成に役立てようとする試みは、今なお活発になされている。天然素材や食品中の生理活性物質の定性については、LC-MSやLC-MS/MSにより、分画することなく対象化合物の同定が可能となるケースが多くなってきてはいるが、これらの手法で化学構造の決定に至らない場合は、対象化合物を単離・精製した後、MSやNMRにより同定(モノ取り・構造決定)されることが多いだろう。最近では、分析技術の発展により、モノ取り・構造決定も、単離品が1 mg以下であっても定性できる時代となり、単離・精製等の操作にかける時間は大幅に短縮されてきている。 2.定量NMR法とは1)定量NMR(qNMR)では有機化合物の構成元素やNMRにおける感度などを考慮して、1H-NMRを利用した方法(1H-qNMR)が広く使用されている。1H-qNMRは、2つの化合物間のシグナル面積強度比が「各化合物のモル濃度×各置換基上の水素数」に比例する原理を利用した定量法である。一般的に、NMRは原子核を対象に測定を行うため、これら2つの化合物は同一の化学構造である必要はない。従って、図1に示すように濃度や純度が明確な1つの標品を内標準物質として用いることにより、内標準物質と測定対象化合物のシグナル面積強度比、水素数、重量(秤取量)等の関係から、様々な測定対象化合物の絶対定量が可能な方法である2, 3)。 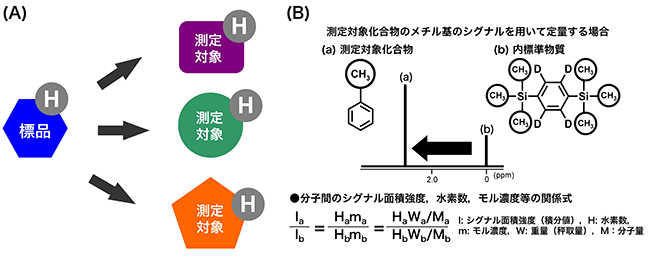 図1 1H-qNMRの原理 1H-qNMRは(A)に示すように原子核を基準にした定量法であるため、1つの標品で化学構造の異なる様々な有機化合物を(B)に示す関係式に基づいて定量することが可能である。 この1H-qNMRで精確な定量を行うためには、シグナル面積強度を正確に得ることが非常に重要である。通常1H-NMRでは、構造解析のために測定の迅速性やS/N(感度)の向上に重点をおいて測定することが多い。しかし、この測定条件では、得られるシグナル面積強度に定量性はなく、定量分析には不向きと言える。SugimotoらやIharaらは、十分なパルス繰り返し時間やデジタル分解能の設定、データ取り込み中の13C核のデカップリングなど、定量用に最適化された測定条件を確立し、その条件を用いた1H-qNMRの実例を報告している4,5)。また、最近では測定結果の計量計測トレーサビリティを確保するため、内標準物質として認証標準物質を用いた1H-qNMRが報告された6)。一般に、市販されている認証標準物質の数は非常に少ないため、測定対象化合物と同一の標準物質を必要とする各種クロマトグラフィーでの利用には限界がある。1H-qNMRでは、その測定原理から、1つの認証標準物質を利用することで様々な測定対象化合物の国家標準へとつながる定量値を得ることができ、定量値の信頼性の向上に大きく貢献できる。このような特徴から、1H-qNMRは、生薬や既存添加物中の主要成分の含量分析7-10)、加工食品中の保存料や甘味料の含量分析11-14)、多環芳香族炭化水素、かび毒、下痢性貝毒等の分析で用いる定量用試薬の純度分析15-17)のほか食品添加物標品や日本薬局方試薬の純度分析など公的な試験法への導入も進んでいる18-21)。 3.単離品への1H-qNMRの利用このように、1H-qNMRは、測定対象化合物の絶対量の算出が可能な方法であり、その分析値は計量学的に正確なため信頼性が高いといえる。そこで、我々は食品成分分析への1H-qNMRの利用に向けた検討の一環として、ゴマ葉中から単離したアクテオシド22) の純度分析を行った。図2に示すように、認証標準物質であるDSS-d6を内標準物質として含有するMeOH-d4/D2O(6:4)を測定溶媒として用いたとき、δ 6.34 ppm(水素数1、シグナル①)、δ 6.66 ppm(水素数1、シグナル②)、δ 6.76 ppm(水素数1)、δ 6.78 ppm(水素数1)、δ 6.87 ppm(水素数1、シグナル③)、δ 7.04 ppm(水素数1、シグナル④)、δ 7.13 ppm(水素数1、シグナル⑤)およびδ 7.63 ppm(水素数1、シグナル⑥)にアクテオシドの芳香族水素に由来するシグナルが観察された。これらは、δ 3~5 ppm付近に観察される糖部の水素に由来するシグナルと比較して、シグナル間の分離度が良好であったことから、定量用のシグナルとして適切と考えられた。しかし、これらの領域の各シグナルの分離度を詳細に観察すると、ヒドロキシチロソール残基の2位(δ 6.76 ppm)と5位(δ 6.78 ppm)に由来するシグナル(図2(B)の②と③の間のシグナル)は、シグナルのすそが部分的に重なっており、適切な積分範囲の設定が困難であることから、定量用シグナルとしては不適と考えられた。 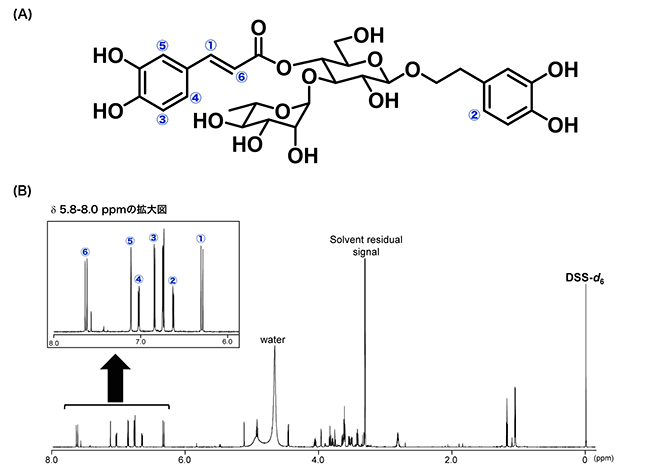 図2 アクテオシドの化学構造(A)および単離したアクテオシドの1H-qNMRスペクトル(B) これらを除く6種のシグナルを用いてアクテオシドの含量を算出したところ、表1に示すように、δ 6.66 ppmのシグナルからの含量値は他の5種のシグナルからの含量値と比較してやや低い値を示したものの、これら6種のシグナルから算出された含量値に大きな違いは認められなかった。
表1 各シグナルより算出された
アクテオシド単離品の含量 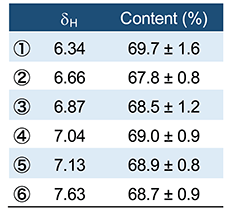 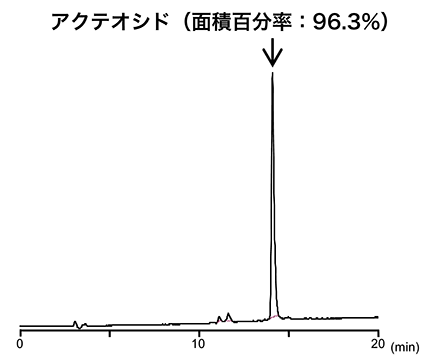 図3 単離したアクテオシドのLCクロマトグラム LC条件、カラム:Waters X-Bridge C18(5μm, 4.6×150 mm)、 移動相:A, 0.1%ギ酸、B, 0.1%ギ酸含有アセトニトリル(0-15 min; 5-35%B, 15-25 min; 35-100%B, 25-26 min; 100%B)、流速:0.8 mL/min、カラムオーブン:40 ℃、検出波長:254 nm。 1H-qNMRスペクトル(図2)では、δ 1.0 ppm、δ 2.0 ppm、δ 5.0~6.0 ppm付近などにアクテオシドの単離・精製の際に使用したメタノール、エタノールなどの溶媒に由来するシグナルや微量な未同定の夾雑成分に由来するシグナルが複数観察されており、それらのシグナルの数や面積強度を考えると、単離されたアクテオシドの含量が少なくとも96.3 %ではないことは明白だろう。なお、我々は市販のアクテオシド標品を用いてLC分析を行い、絶対検量線法によりアクテオシド単離品の定量を行ったところ、その含量値は68.8 %を示し、1H-qNMRの結果と大きな違いは認められなかったことを確認している。このように、単離品の含量分析において、面積百分率法による含量や純度の評価には当然ながら限界があり、定量用標品が存在しない化合物であれば絶対検量線法による含量の算出も不可能である。また、単離品の場合、精製操作で用いた有機溶媒や水が不純物として大きな影響を及ぼす場合も多いが、厳密にこのような不純物の残留量を考慮して測定対象化合物の含量を算出することは多大な時間と労力、さらには測定に必要な試料量の増大をもたらす。一方で、1H-qNMRは比較的少ない試料量で測定対象化合物の正確な定量が可能であることから、特に、貴重な単離品の含量値の測定では本法は非常に有用であり、1H-qNMRから得られた含量値を基に定量分析や活性評価を行えば、データの質の向上に大きく貢献できるだろう。 4.1H-qNMRによる食品中有用成分の定量分析ここまでは、単離した化合物の含量分析に焦点を当ててきたが、1H-qNMRは食品や食品素材などに含まれる有用成分等の分析にも利用が可能と考えられる。一般的に、食品中の有用成分などを分析する際には、抽出、精製、場合によっては濃縮操作などを行った後、GC、LC、LC-MSなどを用いることが多いが、試料によっては煩雑な前処理、低回収率、マトリックス効果等の問題もあり、正確な定量が困難なケースも珍しくない。1H-qNMRは、食品中の食品関連成分の定量において表2に示す特徴と利点をもった分析法と言え、本法を食品分析へ応用すれば、食品の品質や安全性の評価などにおける定量値の信頼性は飛躍的に向上するものと期待される。
表2 食品分析における1H-qNMRの特徴と利点
 我々は、食品中の有用成分の分析における1H-qNMRの応用に関する検討として、2種のゴマ若葉乾燥粉末中のアクテオシドの分析を行った。図4に示したように、カフェ酸残基の2位(シグナル⑤)のシグナルは、アクテオシド由来のシグナルの中で夾雑成分の影響を最も受けにくいシグナルと考えられた。そこで、このシグナルを定量用シグナルとして設定しアクテオシド含量を算出したところ、試料Aは10.1±0.4 %、試料Bは1.1±0.03 %であった。 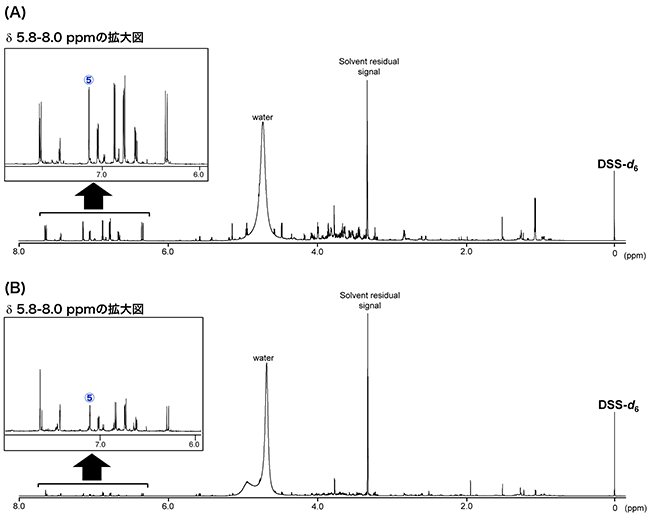 図4 ゴマ若葉乾燥粉末の1H-qNMRスペクトル (A) 試料A、(B)試料B 一方で、これらの試料について、LCで分析し、絶対検量線法により定量したところ、両試料の含量値は1H-qNMRの結果と同等であった(表3)。今回の1H-qNMRでは、ゴマ若葉乾燥粉末をDSS-d6を含有するMeOH-d4/D2O(6:4)に入れ、可溶部をそのまま測定した。測定に際して、煩雑な前処理操作はなく、クロマトグラフィーのような分離操作や検量線の作成なども必要ない。このように、本法は信頼性のみならず、迅速性にも優れた方法であることが実証され、食品や食品素材などの含有成分等の分析にも有用と考えられた。
表3 1H-qNMRおよびLCより算出されたゴマ若葉乾燥粉末中のアクテオシド含量の比較
 5.まとめ1H-qNMRはシグナルの分離機構がクロマトグラフィーとは根本的に異なるため、クロマトグラフィーで分離が困難な化合物についても本法で定量が可能となるケースが期待される。特に、食品のような夾雑成分を多く含む試料では、測定対象化合物を迅速かつ精確に定量できる有用なツールの1つとしてその威力を発揮するだろう。なお、1H-qNMRを行う上で、定量値の精確さを保証するためには、認証標準物質の利用、試料や標準物質の精密な秤量、定量用に最適化されたNMR測定条件の設定、適切な定量用シグナルの選択、十分なシム調整、データ解析時の位相補正、ベースライン補正及び積分範囲の適切な設定などが重要である。これらの点に注意を払いながら1H-qNMRを利用することが望まれる。 参考文献1 大槻崇:化学と生物,52,622(2014). 2 N. Sugimoto, M. Tahara, T. Suematsu & T. Miura: Food Hyg. Saf. Sci., 53, J-228(2012). 3 合田幸広:医薬品医療機器レギュラトリーサイエンス,44,753(2013). 4 N. Sugimoto, A. Tada, T. Suematsu, K. Arifuku, T. Saito, T. Ihara, Y. Yoshida, R. Kubota, M. Tahara, K. Shimizu, S. Ito, T. Yamazaki, Y. Kawamura& T. Nishimura: Food Hyg. Saf. Sci., 51, 19(2010). 5 T. Ihara, T. Saito & N. Sugimoto: Synthesiology, 2, 12(2009). 6 T. Saito, T. Ihara, M. Koike, S. Kinugasa, Y. Fujimine, K. Nose & T. Hirai: Accred. Qual. Assur., 14, 79(2009). 7 K. Hasada, T. Yoshida, T. Yamazaki, N. Sugimoto, T. Nishimura, A. Nagatsu & H. Mizukami: J. Nat. Med., 65, 262(2011). 8 A. Tada, K. Takahashi, N. Sugimoto, T. Suematsu, K. Arifuku, T. Saito, T. Ihara, Y. Yoshida, K. Ishizuki, T. Nishimura, T. Yamazaki & Y. Kawamura: Food Hyg. Saf. Sci., 51, 205(2010). 9 T. Yoshida, K. Terasaka, S. Kato, F. Bai, N. Sugimoto, H. Akiyama, T. Yamazaki & H. Mizukami: Chem. Pharm. Bull, 61, 1264(2013). 10 R. Tanaka, H. Shibata, N. Sugimoto, H. Akiyama & A. Nagatsu: J. Nat. Med., 70, 797(2016) 11 T. Ohtsuki, K. Sato, N. Sugimoto, H. Akiyama & Y. Kawamura: Anal. Chim. Acta, 734, 54(2012). 12 T. Ohtsuki, K. Sato, N. Sugimoto, H. Akiyama & Y. Kawamura: Talanta, 99, 342(2012). 13 T. Ohtsuki, K. Sato, N. Sugimoto & H. Akiyama: Food Chem., 141, 1322(2013). 14 T. Ohtsuki, K. Sato, Y. Abe, N. Sugimoto & H. Akiyama: Talanta, 131, 712(2015). 15 田原麻依子,末松孝子,早川昌子,合田幸広,小西良子,杉本直樹:Mycotoxins,62,111(2012). 16 田原麻依子,杉本直樹,大槻崇,多田敦子,穐山浩,合田幸広,五十嵐良明:環境科学会誌,27,142(2014). 17 T. Kato, M. Saito, M. Nagae, K. Fujita, M. Watai, T. Igarashi, T. Yasumoto & M. Inagaki: Anal. Sci., 32, 729(2016). 18 平成23年厚生労働省告示第307号 19 平成25年厚生労働省告示第45号 20 平成25年厚生労働省告示268号 21 第十七改正日本薬局方 22 松藤寛,大森潤一,後藤修一,千野誠,和田悦治,内田あゆみ,深堀勝謙,山形一雄,櫻井英敏:日本食品科学工学会誌,58,88(2011). 略歴松藤 寛(マツフジ ヒロシ) 大槻 崇(オオツキ タカシ)  サナテックメールマガジンへのご意見・ご感想を〈e-magazine@mac.or.jp〉までお寄せください。 |
| Copyright (C) Food Analysis Technology Center SUNATEC. All Rights Reserved. |